Johns Hopkins APL Technical Digest
Assessment of Sequencing for Pathogen-Agnostic Biothreat Diagnostics, Detection, and Actionability for Military Applications

Christopher E. Bradburne, Robert A. Player, Sarah L. Grady, Ellen R. Forsyth, Kathleen J. Verratti, and Jeffrey B. Bacon
Abstract
Biothreat detection strategies have historically focused on cheap, specific, and deployable assays that detect a small but specific nucleic acid or protein component of a threat organism. Genomic sequencing technologies that have emerged over the past 15 years are poised to find their place in the biothreat detection tool kit for military and civilian use. Here we describe efforts to compare and contrast sequencing to traditional polymerase chain reaction (PCR) assays for diagnostics and detection of biothreat agents of concern in military applications. We show that after direct spiking of human blood and serum with biothreat simulants, agnostic sequencing can achieve detection. However, for known agents, PCR is still superior in terms of speed, cost, scale, and reliability for military applications. Although PCR should still be the first choice for diagnostics and detection when an agent is known or suspected, for unknown agents, agnostic sequencing can be a powerful addition to identify causative agents in soils, aerosols, and biothreats in patient samples. APL developed and conducted this work for the Department of Defense to address the basic question of when to use PCR versus when to use sequencing for field-forward infectious disease diagnostics and environmental detection.
Download this article or scroll to continue reading
Introduction
Over the past 30 years, rapid biological threat detection and identification has been enabled by small, mobile, cheap, and specific polymerase chain reaction (PCR) assays for DNA and RNA1 and lateral flow assays (LFAs) for proteins and antigens.2 The advantage of PCR for detection is that it can be made to target DNA sequences that are specific to a pathogen while also excluding closely related, but nonpathogenic microbes and viruses (i.e., “near neighbors”). PCR enables enzymatic amplification from, in theory, a single DNA molecule to millions of copies. This allows the target signal to be amplified from very low levels relative to background noise. In addition, the abundance of pathogen target can be estimated based on how long it takes the signal to cross a threshold of detection (termed a cycle threshold, or Ct, value) in a quantitative PCR (qPCR) assay. A Ct value can therefore constitute both a detection signal and an abundance signal, with lower values indicating higher amounts of initial material.
Because PCR is a moderately complex molecular assay, an automated PCR platform (FilmArray) that can run several targeted pathogens is currently used across the military. A simpler, hand-held option is the LFA. An LFA is a paper-based chromatography assay that uses the wicking properties of paper to separate sample components and then expose biothreat target proteins or antigens to a detection antibody. Those antibodies are linked to a chromatographic indicator that gives either a positive or negative indication if the antibody comes into contact with the antigen. LFAs are simple and robust and include such widespread applications as home pregnancy tests. As such, they are excellent for quick answers but can struggle with sensitivity and low-abundance target samples.
Genomic sequencing involves assessing a sample for the genomic content and the specific sequence of nucleotides of each genomic fragment in that sample. Because early Sanger sequencing could sequence only one DNA fragment at a time, throughput, scale, time, and cost made it untenable for field-forward diagnostics. The field changed in 2007 with the commercial development of a massively parallel genome sequencer able to generate millions of sequencing reads of varying lengths. Since then, a handful of technology companies have pushed the market forward.3,4 Their products vary in terms of ease of use, lengths of individual DNA reads, per-base quality, and throughput. The cost of sequencing per base has also been driven down by at least four orders of magnitude since 2007.5
Despite these advances, sequencing still has not made it into widespread use in field-forward disease diagnostics or in environmental biothreat detection for military applications. The major barriers are the cost and complexity of individual sequencing runs and difficulties in analyzing and interpreting results. All sequencing technologies produce immense amounts of data (on the order of a hundred megabytes to hundreds of gigabytes), and the analysis can typically be done only on highly capable laptops, the cloud, or high-performance computers. Considerable technical knowledge and skill are required to perform bioinformatics, and interpretation can be difficult even when automated pipelines are employed. Examples of bioinformatic complexity include the presence of many near-neighbor sequences in a sample, faulty and mis-curated reference genomes, and differences in the abundance of the target versus background that result in the target not being detected. Because these and other challenges have been difficult to solve, the US Food and Drug Administration (FDA) has not fully approved genomic sequencing technologies for diagnostic use, even though it issued “draft guidance” for the industry in 2016 with the expectation that sequencing for diagnostics would eventually be approved.6,7 To our knowledge, there has been no updated guidance since this draft document was released.
One of the simple questions the Department of Defense (DoD) seeks to answer is when to employ LFA and/or PCR-type technologies and when to employ genomic sequencing technologies. In 2019, just before the start of the COVID-19 pandemic, the Defense Threat Reduction Agency (DTRA) tasked APL to address this question, using data and experience to guide DoD stakeholders on when, where, why, and how to utilize this emerging capability. Partners at the US Army Medical Research Institute of Infectious Diseases (USAMRIID) had developed and/or adopted three protocols employing different methodologies to enrich viral sequences in metagenomic samples so that they could be detected, counted, and characterized:
1. Sequence-independent, single-primer amplification (SISPA)8 employs single primers targeting a virus, with random hexamers to allow virus taxa amplification without knowledge of the viral genome beyond the single target primer. This allows a mostly sequence-agnostic enrichment of the virus in a sample undergoing next-generation sequencing (NGS).
2. Sequence-independent, single-primer amplification, and rapid amplification of cDNA ends (SISPA-RACE)8 employs SISPA, but includes rapid amplification of the cDNA ends after reverse transcription.
3. Hybrid oligonucleotide enrichment amplification9 involves utilizing bioinformatics optimization to select many primer oligonucleotide sequences to enrich for a variety of viruses.
A challenge in agnostic diagnostic sequencing is determining which strategy to select to optimize the chances of detection. In true field-forward settings, field personnel may be able to draw or obtain diagnostic samples, but they may not know whether to target bacteria, viruses, or even fungi as the causative agent. Of these agents, viruses are typically the most difficult to detect using NGS because of their small genome size relative to the host and the fact that they may not be present in high titers in a clinical sample. Therefore, employing a sequencing protocol to enrich viruses would enable virus detection while still generating enough sequencing reads to detect any bacterial pathogens present. With this concept in mind, APL designed a test bed to evaluate these viral-enrichment, yet pathogen-agnostic, sequencing pipelines. The data generated gave the DoD important insights into the performance of sequencing versus PCR, and one of the protocols, hybrid enrichment, was a forerunner of the ARTIC protocol that clinical laboratories and researchers used to enrich and sequence SARS-CoV-2 viruses from clinical diagnostic samples during the COVID-19 pandemic.10
Approach
Biothreat Diagnostics
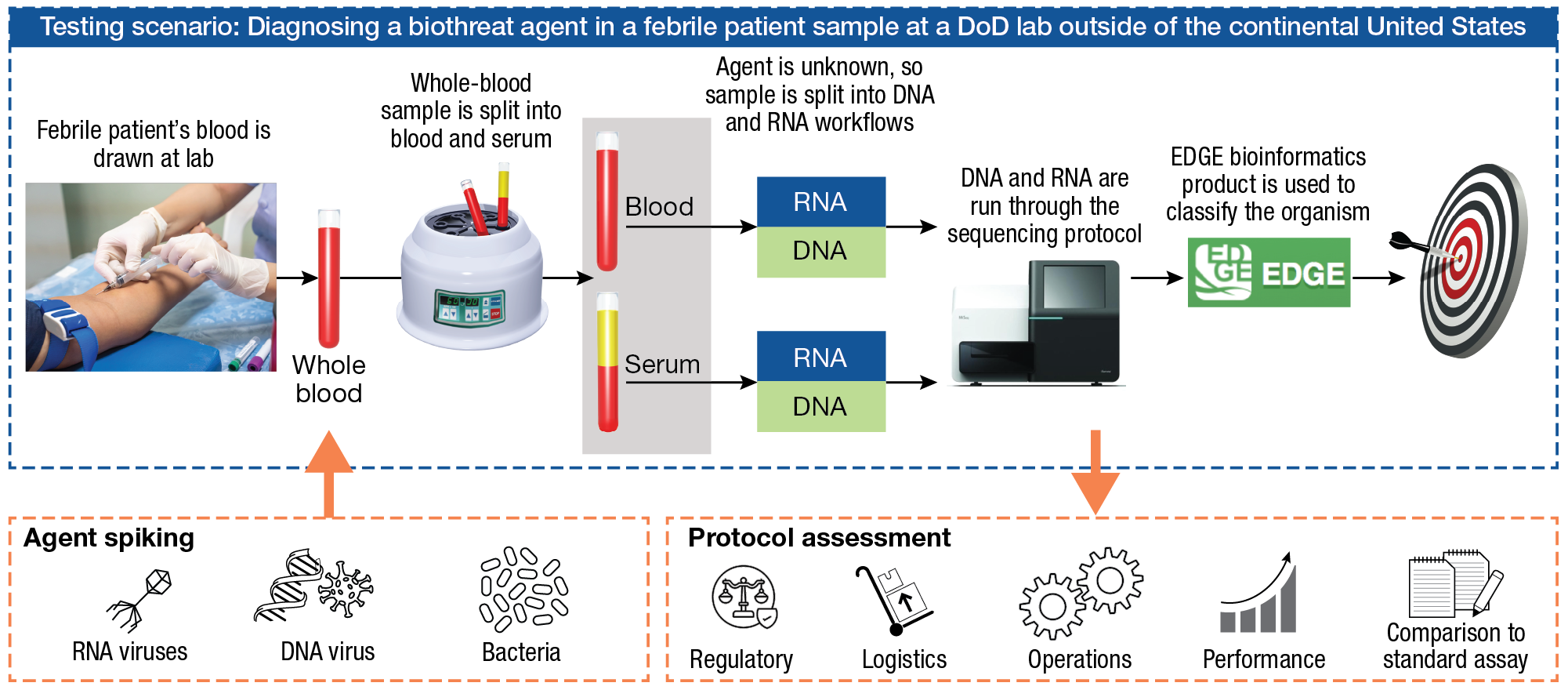
For testing, APL designed a scenario-based test that envisions a febrile patient infected with an unknown biothreat agent (Figure 1). The patient provides a blood sample, which is divided into whole blood and serum. This biothreat scenario is applicable to many potential select agents that achieve bacteremia/viremia at or beyond the febrile phase, but other clinical specimens could also be appropriate. (“Select agents are biological agents and toxins that have been determined to have the potential to pose a severe threat to public health and safety, to animal and plant health, or to animal or plant products.”11) In this scenario, since the causative agent is not known, each sample is probably split by a lab technician for DNA and RNA processing and then entered into the NGS protocol. After sequencing, the sample is analyzed using a DTRA-funded analysis software, Empowering the Development of Genomics Expertise (EDGE) Bioinformatics, to determine the presence and abundance of an agent. APL was the project integration lead involved in initiating the development of this software with Los Alamos National Laboratory and the Naval Medical Research Command in 2013. To test this pipeline against an array of pathogen genomes, four Biosafety Level (BL) 2 or BL3 agents are used: Bacillus anthracis Ames (Ba), a bacterium; vaccinia virus (VV), a double-stranded (ds) DNA virus; Venezuelan equine encephalitis virus (VEEV), a (+) single-stranded (ss)RNA virus; and Seoul hantavirus (Ha), a (–) ssRNA virus. Each agent is tested with each sequencing protocol, as well as with agent-specific qPCR assays, at a concentration in blood or serum corresponding to human clinical concentrations reported in the literature (Table 1). This helps to ensure some fidelity of the use case of a febrile patient who might appear with an infection from that pathogen.
Figure 1
APL testing scenario for comparison of pathogen-agnostic sequencing assays for use in field-forward diagnostics. The use case is a febrile patient who walks into a forward, remote, low-resource clinic. An agnostic sequencing approach is initiated to look for the presence of RNA and DNA viruses and bacteria. Sequencing protocols are assessed against the PCR standard assay for performance, ease of use, cost, speed, and regulatory considerations.
| Table 1. Pathogens chosen for spiking blood and serum and the corresponding human clinical disease concentrations targeted | ||||||
| Organism in the Literature | Equivalent Test Organism | Host | Range of Clinical Concentration | Measured in | Reference | Concentration Chosen for Testing |
| Bacillus anthracis Ames ancestor | Ba | African green monkeys | 40 to > 1e3 CFU/mL | Blood | Rossi et al.12 | 1e5 CFU/mL |
Sin Nombre (Hanta) Puumala (Hanta) | HV | Human | 1e4.5 to 1e7.5 PFU/mL 3 to 1.8e6 PFU/mL | Blood Serum | Terajima et al.13 Evander et al.14 | 1.1e3 PFU/mL |
| Venezuelan equine encephalitis virus | VEEV | Human | 1e5 to 1e7 PFU/mL 1e2 to 1.8e4 PFU/mL 3e2 to 6.7e5 PFU/mL 1e1.7 to 1e1.56 PFU/mL | Blood Serum | Sellon and Long15 Vilcarromero et al.16 Quiroz et al.17 Weaver et al.18 | 7.5e3 PFU/mL |
Variola virus Variola virus Monkeypox | VV | Macaques | Up to 1e4 PFU/mL Up to 2e7 genomes/mL Up to 4.8e6 genomes/mL | PBMC Blood Blood | Rubins et al.19 Mucker et al.20 Barnewall et al.21 | 1e5 PFU/mL |
| PBMC, peripheral blood mononuclear cell. | ||||||
To support application to real-world scenarios, all protocols were evaluated for cost and operational time of use. Lastly, even though the scenario was for a use case outside the continental United States, there was an interest in mapping to regulatory requirements for the use of diagnostic tests in US jurisdictions. FDA regulatory oversight of sequencing for clinical diagnostics has been challenging because sequencing is so sensitive and can reveal so much information that the results can often be difficult to interpret. For example, nearly every metagenomic sample will have sequence reads that map to pathogens, and yet those pathogens may not constitute a threat for a number of reasons: they may be identical to nonpathogenic near-neighbor organisms, associated with nonviable threats, or associated with nonvirulent variants of an infectious agent, or they may constitute pathogenic sequences that appear across many different microbial taxa. As mentioned earlier, the FDA’s draft regulatory guidance provides a comprehensive assessment of a diagnostic test using sequencing, in anticipation of eventual approval, but as of this publication date, there is still no official FDA approval of sequencing for infectious disease diagnostics as described here. Nevertheless, we compared the operation of these three protocols with the draft FDA guidance document.
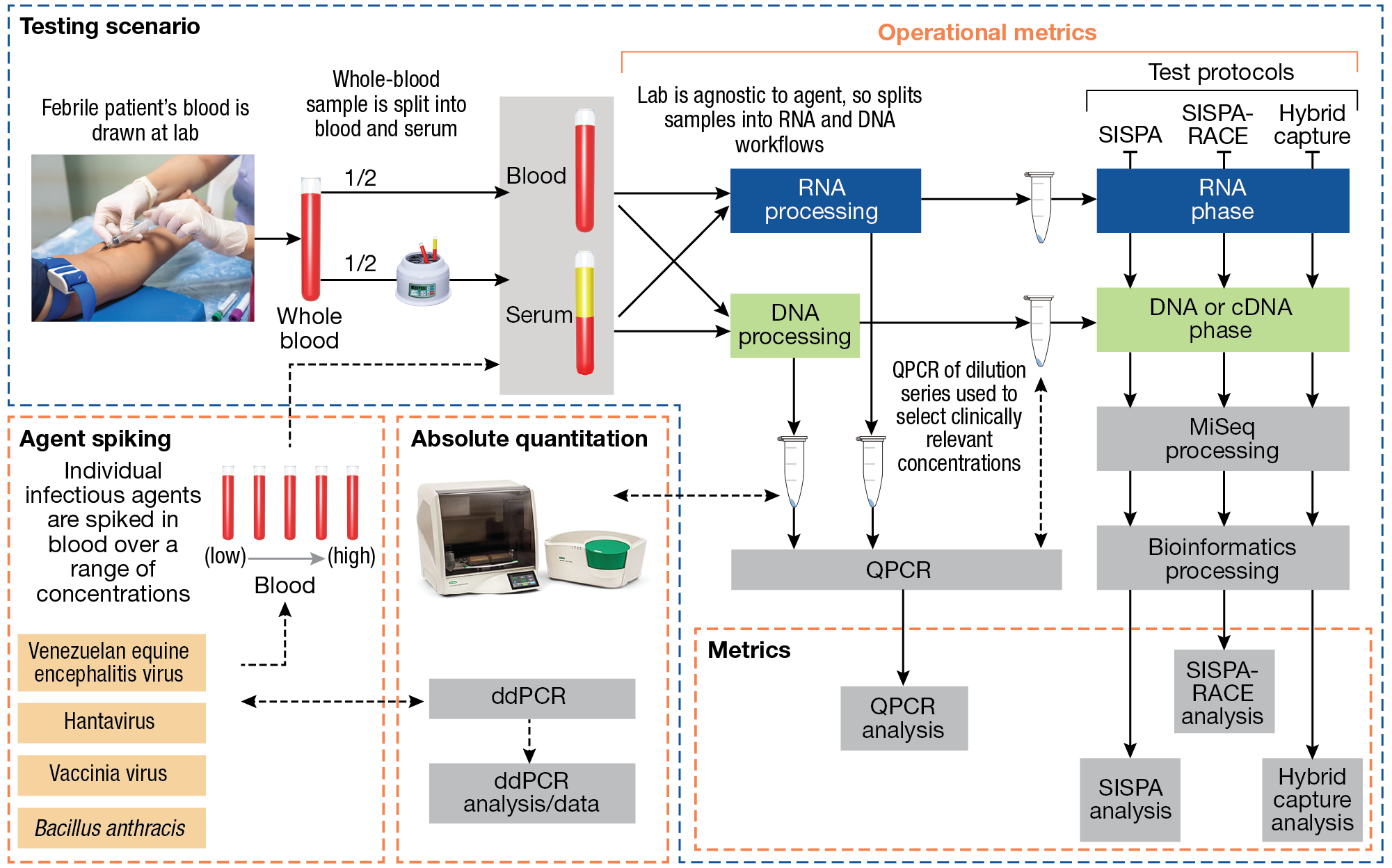
Performance metrics for each protocol are shown in Figure 2. Each protocol was evaluated using spiked agent from blood and serum and compared directly with qPCR and droplet-digital PCR (ddPCR) for extensive quantification. This enabled direct comparison of the protocols’ performance and sensitivity.
Figure 2
Compartmentalization of performance metrics and process quality control for the APL diagnostic sequencing test scenario. Spiked pathogen concentrations are confirmed by droplet-digital PCR (ddPCR) and qPCR.
Fielded Biothreat Detection
Aiming to put sequencing technologies in the hands of service members in far-forward environments, DTRA has funded efforts to transition a small, portable sequencing capability to special operations teams. APL has supported components of these efforts by providing subject matter expertise and participating in exercises demonstrating these capabilities. In February 2024, DTRA and the US Army Combat Capabilities Development Command Chemical Biological Center (DEVCOM CBC) hosted a testing and training exercise just south of the Arctic Circle in Fox, Alaska, to support testing and evaluation for the Army’s Far Forward Advanced Sequencing Technology (F-FAST), led by Dr. R. Cory Bernhards of DEVCOM CBC. F-FAST, which has now transitioned into the Far Forward Bio-detection System (FFBS), uses an Oxford Nanopore platform called the M1kC, a handheld device that contains a readout display and flow cell cartridge. Supporting materials for sample and library preparation are labeled in a kit that is deployed with the M1kC. This system is designed to agnostically provide the genetic content of a noncomplex sample and information about a potential threat agent in under an hour for microbial pathogens and DNA viruses and in 90 min for RNA-based viruses. At the time of the exercise, the F-FAST system had the ability to identify known biological pathogens by using a custom database containing the full genomes of ~5,000 organisms.
The F-FAST system has already been tested and evaluated in multiple environments, including Dugway Proving Ground during the peak of summer when temperatures were over 100°F. This exercise sought to test performance in the extreme cold at Fort Wainwright, Alaska, home to the Army’s 11th Airborne Division “Arctic Angels.” The division’s mission is to conduct expeditionary operations within the Indo-Pacific theater, but it is specialized to conduct multi-domain operations in the Arctic.22
Results
Biothreat Diagnostics
The results of the scenario testing support the general idea that when the agent being tested for is known, qPCR—and not sequencing—should be used. Only cases with an unknown causative agent could begin to justify the cost and time required to implement sequencing to detect microbial and viral infections. Table 2 demonstrates the significant cost and time to obtain results for the Illumina-based sequencing protocols tested. All protocols took between 88.2 and 107.3 h to reach a result, and the minimum materials cost was $1,730. In contrast, qPCR took only 4.25 h to reach a result on average, while costing only $420. Furthermore, this qPCR cost estimate is probably high, as the materials would generally be bought and implemented at scale, whereas this was a single-usage test.
| Table 2. Time and cost per answer for sequencing vs. qPCR | |||
| Sequencing Protocol or qPCR | Source | Time to Answer (h) | Materials Cost per Answer ($) |
| Nextera XT | Illumina | 92.85 | 2,906 |
| SISPA | USAMRIID | 88.5 | 3,069 |
| SISPA-RACE | USAMRIID | 88.2 | 1,730 |
| Hybrid Capture | USAMRIID | 107.3 | 2,352 |
qPCR performance was also excellent, with zero false positives and nominal detection of each organism consistent with the amount of spiked material (Table 3). This supports the case for qPCR to continue to be the gold standard when using the diagnostic test to confirm or deny the presence of a known agent.
| Table 3. qPCR performance for detection of spiked organisms | |||
| Target Organism | Spiked Organism Concentration (PFU/mL) | Ct Value | |
| Whole Blood | Serum | ||
Bacillus anthracis Ames ancestor BA-negative control | 1.00E + 05 0.00E + 00 | 27.8 0 | 28.7 0 |
Vaccinia virus Wyeth VV-negative control | 1.00E + 05 0.00E + 00 | 23.1 0 | 22.9 0 |
Venezuelan equine encephalitis virus TC-83 VEEV-negative control | 7.50E + 03 0.00E + 00 | 27.2 0 | 24.0 0 |
Seoul hantavirus Baltimore HA-negative control | 1.10E + 03 0.00E + 00 | 29.6 0 | 29.6 0 |
The sequencing tests generally performed well, but some conflicting results made interpretation difficult. As shown in Table 4, all sequencing protocols detected the Bacillus anthracis Ames ancestor bacteria, generating no false positives or false negatives. SISPA and SISPA-RACE failed to detect the vaccinia dsDNA virus infectious agent in both whole blood and serum. For VEE, the ssRNA (+) virus, SISPA was adequate, but SISPA-RACE failed to detect the virus in whole blood. The most difficult taxon to detect was clearly the ssRNA (–) hantavirus. This pathogen typically grows to low titers in the lab and in infections, so it was expected to be difficult to detect. Hantavirus reads were detected in the SISPA-RACE protocol, but not enough to fall above the threshold of the EDGE bioinformatic filter that makes the decision to call the organism present in the sample (data not shown).
Across all tested protocols, the hybrid capture protocol performed the best and failed to detect only the hantavirus. This was not surprising because, as noted above, hantavirus typically grows to low titers in clinical infections and can be difficult to detect in clinical samples. The ability to enrich viral targets in large amounts of background was a core strategy of the ARTIC protocol that was used to detect SARS-CoV-2 and improved throughout the pandemic, allowing whole-genome modeling of geotemporal viral dispersion by variants in human populations throughout the world.23
| Table 4. Performance of agnostic sequencing workflow without enrichment (Nextera XT) and with the three enrichment protocols (SISPA, SISPA-RACE, and Hybrid Capture) | |||||||||
| Protocol | Database | Target Organisms | |||||||
| gram+ bacteria: Bacillus anthracis Ames ancestor | dsDNA virus: Vaccinia virus Wyeth | ssRNA (+) virus: Venezuelan equine encephalitis virus TC-83 | ssRNA (–) virus: Seoul hantavirus Baltimore | ||||||
| Whole Blood | Serum | Whole Blood | Serum | Whole Blood | Serum | Whole Blood | Serum | ||
| Nextera XT | Bacterial | TP,TN,TP | TP,TN,TP | TN,TN,TN | TN,FP,TN | FN,TN,– | TN,TN,– | TN,TN,– | TN,TN,– |
| Viral | TN,TN,TN | TN,TN,TN | TP,TN,TP | TP,TN,TP | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,– | |
| SISPA | Bacterial | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,TN | TN,TN,TN | TN,TN,– | TN,TN,– |
| Viral | TN,TN,– | TN,TN,– | FN,TN,– | FN,TN,– | TP,TN,TP | TP,TN,TP | FN,TN,– | FN,TN,– | |
| SISPA-RACE | Bacterial | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,TN | TN,TN,TN | TN,TN,– | TN,TN,– |
| Viral | TN,TN,– | TN,TN,– | FN,TN,– | FN,TN,– | FN,TN,TP | TP,FP,TP | FN,TN,– | FN,TN,– | |
| Hybrid capture | Bacterial | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,– | TN,TN,TN | TN,TN,TN | TN,TN,– | TN,TN,– |
| Viral | TN,TN,– | TN,TN,– | TP,TN,– | TP,FP,– | TP,TN,TP | TP,TN,TP | FN,TN,– | FN,TN,– | |
| The data in each square represent (1) the spiked sample, (2) the negative control, and (3) the positive control. For example, TP,TN,TP indicates that the spiked sample is detected (true positive), the negative control is undetected (true negative), and the positive control is detected (true positive). Green shading, expected result; yellow shading, conflicting result; red shading, false positive result; dashed red outline, non-scenario-linked result. | |||||||||
Discussion
These results clearly show the difference between utilization of PCR for targeting known pathogens versus utilization of sequencing to characterize an unknown organism. Generally, if the target organism is known, the best guidance is the gold standard of PCR. PCR is cheap, timely, and reliable, and its easily interpretable results minimize false positives. However, a negative result may lead to more questions than answers, leaving the operator without a diagnostic call and yet still with a febrile patient with a likely infectious disease. In this case, sequencing with the hybrid enrichment protocol described in this article offers the best opportunity to detect a causative agent. In addition, using the protocol offers the best chance to obtain sequence-based information that could indicate whether the agent is a new strain or variant or, in the case of a microbe, whether there is any antimicrobial resistance, as well as any other countermeasure-relevant information present.
The information payoff gained from sequencing rather than PCR is apparent, and in many cases, even a positive PCR result could be important for confirmation. One intriguing option is direct sequencing of the amplicons from a PCR result. Through support and direction from the Defense Biological Product Assurance Office (DBPAO), APL has developed a methodology to do exactly this. An APL team modified biothreat-specific PCR primers to add library adapter sequences so that the amplicon product could be directly inserted into the Oxford Nanopore sequencing protocol. The resulting approach allows a user to run a PCR assay for a biothreat agent and quickly confirm via sequencing the agent and its abundance.24
Biothreat Detection in Cold Regions
Work is ongoing to package the hybrid detection technology into a handheld, field-portable sequencing-based viral detection capability. It was first demonstrated during the Ebola outbreak in West Africa just prior to the COVID-19 pandemic in 2019.25 Commercial products offer handheld sequencers and informatics hardware. Computational power is limited, but future models of field-portable sequencing devices could overcome these limitations. These products also have the potential to be utilized in adverse environments, such as in subfreezing conditions. In February 2024, APL attended a DTRA-supported US Army Special Operations Command (SOCOM) demonstration of a handheld sequencer in arctic conditions at the Cold Regions Research and Engineering Laboratory (CRREL) permafrost tunnel facility in Fox, Alaska (Figure 3). During the 5 days of testing, temperatures ranged from 15°F to –25°F. During the first few runs of these exercises, operators had difficulty correctly loading the sequencer flow cells without adding disruptive air bubbles that confounded results. In addition, the cold weather dictated innovations such as using body heat to keep reagents from freezing before use. By the last 2 days, however, the soldiers were able to operate the sequencer and achieve the expected results nominally (data not shown). More information on the exercise is available in CBNW Magazine.26 The exercise illustrated that the technology could be used in a training scenario in cold environments with several modest adaptations.
Figure 3
Field demonstration of field-forward sequencing for biothreat detection. This demonstration took place at the February 2024 Arctic Edge exercise facilitated by the US Northern Command at the CRREL permafrost tunnel facility in Fox, Alaska. During the exercise, US Army Special Operations Command demonstrated use of a handheld sequencer developed by DEVCOM CBC through the F-FAST program.

Conclusion
Robust, deployable diagnostics are an important component of a military unit’s medical surveillance and biothreat detection capability. A central question in field-forward, molecular diagnostics and detection is when to use qPCR versus when to use sequencing, particularly in a far-forward environment and in wide-ranging environmental conditions. The work described here illustrates that for suspected (i.e., known) pathogens, qPCR is still superior to sequencing in terms of cost, complexity, and time to answer. For novel outbreaks, or orthologous confirmation of a new outbreak, agnostic and pathogen-enrichment sequencing has significant utility. In addition, the field demonstration by DEVCOM CBC and SOCOM in the extreme cold and the ability to deploy sequencing to remote locations also offers important advantages for molecular detection of biothreats in austere environments. As costs decrease and ease of use increases, the utility of sequencing should continue to increase.
Acknowledgments: Distribution Statement A—Approved for public release; distribution is unlimited.
- S. Yang and R. E. Rothman, “PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings,” Lancet Infect. Dis., vol. 4, no. 6, pp. 337–348, 2004, https://doi.org/10.1016/S1473-3099(04)01044-8.
- H. R. Boehringer and B. J. O’Farrell, “Lateral flow assays in infectious disease diagnosis,” Clin. Chem., vol. 68, no. 1, pp. 52–58, 2022, https://doi.org/10.1093/clinchem/hvab194.
- C. E. Bradburne, “Personalizing environmental health for the military—Striving for precision,” in Total Exposure Health: An Introduction, K. A. Phillips, D. P. Yamamoto, and L. Racz, Eds., 1st ed., Boca Raton, FL: CRC Press, 2020, https://doi.org/10.1201/9780429263286.
- C. Bradburne, D. Graham, H. M. Kingston, R. Brenner, M. Pamuku, and L. Carruth, “Overview of ‘omics technologies for military occupational health surveillance and medicine,” Mil. Med., vol. 180, suppl. 10, pp. 34–48, 2015, https://doi.org/10.7205/MILMED-D-15-00050.
- K. A. Wetterstrand, “DNA sequencing costs: Data,” National Human Genome Research Institute Genome Sequencing Program, https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data.
- F. Luh and Y. Yen, “FDA guidance for next generation sequencing-based testing: Balancing regulation and innovation in precision medicine,” npj Genom. Med., vol. 3, art. 28, 2018, https://doi.org/10.1038/s41525-018-0067-2.
- “Infectious disease next generation sequencing based diagnostic devices: Microbial Identification and detection of antimicrobial resistance and virulence markers — Draft guidance for industry and Food and Drug Administration staff,” US Food and Drug Administration, May 13, 2016, https://public4.pagefreezer.com/content/FDA/16-12-2022T08:12/https:/www.fda.gov/media/98093/download.
- D. H. Song, W.-K. Kim, S. H. Gu, D. Lee, J.-A. Kim, et al., “Sequence-independent, single-primer amplification next-generation sequencing of Hantaan virus cell culture-based isolates,” Amer. J. Trop. Med. Hyg., vol. 96, no. 2, pp. 389–394, 2017, https://doi.org/10.4269/ajtmh.16-0683.
- J. Quick, N. D. Grubaugh, S. T. Pullan, I. M. Claro, A. D. Smith, et al., “Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples,” Nat. Protoc., vol. 12, pp. 1261–1276, 2017, https://doi.org/10.1038/nprot.2017.066.
- A. W. Lambisia, K. S. Mohammed, T. O. Makori, L. Ndwiga, M. W. Mburu, et al., “Optimization of the SARS-CoV-2 ARTIC network V4 primers and whole genome sequencing protocol,” Front. Med., vol. 9, art. 836728, 2022, https://doi.org/10.3389/fmed.2022.836728.
- Centers for Disease Control and Prevention Office of Readiness and Response, “Division of Regulatory Science and Compliance: What is a select agent?,” https://www.cdc.gov/orr/dsat/what-is-select-agents.htm (last reviewed October 2, 2023).
- C. A. Rossi, M. Ulrich, S. Norris, D. S. Reed, L. M. Pitt, and E. K. Leffel, “Identification of a surrogate marker for infection in the African Green monkey model of inhalation anthrax,” Infect. Immun., vol. 76, no. 12, pp. 5790–5801, 2008, https://doi.org/10.1128/IAI.00520-08.
- M. Terajima, J. D. Hendershot III, H. Kariwa, F. T. Koster, B. Hjelle, et al., “High levels of viremia in patients with the Hantavirus pulmonary syndrome,” J. Infect. Dis., vol. 180, no. 6, pp. 2030–2034, 1999, https://doi.org/10.1086/315153.
- M. Evander, I. Eriksson, L. Pettersson, P. Juto, C. Ahlm, et al., “Puumala hantavirus viremia diagnosed by real-time reverse transcriptase PCR using samples from patients with hemorrhagic fever and renal syndrome,” J. Clin. Microbiol., vol. 45, no. 8, pp. 2491–2497, 2007, https://doi.org/10.1128/jcm.01902-06.
- D. C. Sellon and M. T. Long, Equine Infectious Diseases, 2nd ed., St. Louis, MO: Elsevier, 2014.
- S. Vilcarromero, P. V. Aguilar, E. S. Halsey, A. Laguna-Torres, H. Razuri, et al., “Venezuelan equine encephalitis and 2 human deaths, Peru,” Emerg. Infect. Dis., vol. 16, no. 3, pp. 553–556, 2010, https://doi.org/10.3201/eid1603.090970.
- E. Quiroz, P. V. Aguilar, J. Cisneros, R. B. Tesh, S. C. Weaver, “Venezuelan equine encephalitis in Panama: Fatal endemic disease and genetic diversity of etiologic viral strains,” PLoS Negl. Trop. Dis., vol. 3, no. 6, art. e472, 2009, https://doi.org/10.1371/journal.pntd.0000472.
- S. C. Weaver, R. Salas, R. Rico-Hesse, G. V. Ludwig, M. S. Oberste, J. Boshell, et al., “Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America,” Lancet, vol. 348, no. 9025, pp. 436–440, 1996, https://doi.org/10.1016/S0140-6736(96)02275-1.
- K. H. Rubins, L. E. Hensley, P. B. Jahrling, A. R. Whitney, T. W. Geisbert, et al., “The host response to smallpox: Analysis of the gene expression program in peripheral blood cells in a nonhuman primate model,” Proc. Natl. Acad. Sci., vol. 101, no. 42, pp. 15190–15195, 2004, https://doi.org/10.1073/pnas.0405759101
- E. M. Mucker, A. J. Goff, J. D. Shamblin, D. W. Grosenbach, I. K. Damon, et al., “Efficacy of tecovirimat (ST-246) in nonhuman primates infected with variola virus (smallpox),” Antimicrob. Agents Chemother., vol. 57, no. 12, 2013, pp. 6246–6253, https://doi.org/10.1128/aac.00977-13.
- R. E. Barnewall, D. A. Fisher, A. B. Robertson, P. A. Vales, K. A. Knostman, and J. E. Bigger, “Inhalational monkeypox virus infection in cynomolgus macaques,” Front. Cell. Infect. Microbiol., vol. 2, art. 117, 2012, https://doi.org/10.3389/fcimb.2012.00117.
- US Army 11th Airborne Division website, https://11thairbornedivision.army.mil (accessed Mar 1, 2024).
- J. Merlet, J. Lagergren, V. M. Vergara, M. Cashman, C. Bradburne, et al., “Data-driven whole-genome clustering to detect geospatial, temporal, and functional trends in SARS-CoV-2 evolution,” in PASC ‘23: Proc. Platform for Adv. Sci. Comput. Conf., June 2023, art. 26, pp. 1–7, https://doi.org/10.1145/3592979.3593425.
- R. Player, K. Verratti, A. Staab, C. Bradburne, S. Grady, et al., “Comparison of the performance of an amplicon sequencing assay based on Oxford Nanopore technology to real-time PCR assays for detecting bacterial biodefense pathogens,” BMC Genom., vol. 21, art. 166, 2020, https://doi.org/10.1186/s12864-020-6557-5.
- E. Kinganda-Lusamaki, A. Black, D. B. Mukadi, J. Hadfield, P. Mbala-Kingebeni, et al., “Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, Democratic Republic of the Congo,” Nat. Med., vol. 27, pp. 710–716 , 2021, https://doi.org/10.1038/s41591-021-01302-z.
- J. P. Lee, “Polar pathogens,” CBNW Magazine, Sep. 15, 2024, http://nct-cbnw.com/polar-pathogens/.
Christopher E. Bradburne is a biologist in APL’s Asymmetric Operations Sector. He has a B.S. in biology and a B.S. in biochemistry from Virginia Polytechnic Institute and State University (Virginia Tech), an M.S. in biochemistry from Clemson University, and a Ph.D. in bioscience from George Mason University. He has experience in genomics, personalized medicine, biodefense and infectious disease, nanotechnology, and astrobiology projects. His genomics and personalized medicine experience includes managing clinical utility studies collecting and using personal genomic information for preventative and precision care, as well as applying sequencing technologies to clinical diagnostics, environmental genomic surveillance, and national security applications.
Robert A. Player is a bioinformatician. He previously worked in the Sequencing and Computational Biology Section in APL’s Asymmetric Operations Sector.
Sarah L. Grady is a biologist in APL’s Asymmetric Operations Sector. She has a B.S. in biology from St. Mary’s College of Maryland and an M.S. and a Ph.D. in molecular biology from Princeton University. She has over 12 years of experience in a wide variety of virological, molecular, and microbiology techniques with an emphasis in classical molecular biology/microbiology, virology, synthetic biology, field-forward polymerase chain reaction (PCR) sensors, and -omics technologies (RNA-seq, ribosome profiling, metabolomics, proteomics, etc.).
Ellen R. Forsyth is a molecular biologist in APL’s Asymmetric Operations Sector. She has a B.S. in biology and a B.S. in chemistry from Pennsylvania State University and an M.S. in biotechnology from American University. She is pursuing a post-master’s certificate in sequence analysis and genomics from Johns Hopkins University. She has an extensive background in next-generation sequencing (NGS) and sample preparation and experience in polymerase chain reaction (PCR), clustered regularly interspaced short palindromic repeats (CRISPR), nucleic acid extractions, and other molecular techniques. She is also interested in high-containment sequencing and advanced molecular detection methods.
Kathleen J. Verratti is a molecular biologist in APL’s Asymmetric Operations Sector. She has a B.S. in psychology from Catholic University of America. She has an extensive background in genomics and next-generation sequencing technologies, and her focus has been working with and developing the latest next-generation sequencing technology to solve hard problems. She is the APL project manager for a Defense Biological Product Assurance Office (DBPAO)-funded project focused on applying sequencing technologies to biothreat detection assays.
Jeffrey B. Bacon is a science and technology intelligence professional in APL’s Asymmetric Operations Sector. He holds a B.S. in environmental science from Norwich University; master’s degrees in biology (Norwich University), biotechnology (Air Force Institute of Technology), and engineering management (Missouri University of Science and Technology); and a Ph.D. in international relations (Tufts University). Jeff is an expert in counterproliferation and biosecurity and has served at the highest levels, including leadership roles in the Department of Defense’s response planning and policy implementation during the early phases of the COVID-19 pandemic. In addition to his extensive operational experience, he is also an associate professor and graduate educator at the National Intelligence University.